A.N. Davies
External Professor, University of Glamorgan, UK, Director, ALIS Ltd, and ALIS GmbH – Analytical Laboratory Informatics Solutions
“The man who gets the most satisfactory results is not always the man with the most brilliant single mind, but rather the man who can best coordinate the brains and talents of his associates.”—W. Alton Jones
Background
There has been much debate about which program can predict NMR spectra the best. It is well known within the NMR community that spectra prediction strongly depends on the “quality” of the starting data sets for those systems which use real data as a knowledge base. It has become a hot topic in some blogs, although disappointingly most of the authors tend to have affiliations to one software vendor or another.
Predictions on the whole need to be much more accurate in 1H NMR spectroscopy as opposed to 13C NMR in order to help the scientist. In general the prediction software available to date does a pretty good job when the results of the predictions are used sensibly by well-trained and experienced spectroscopists who recognise the pitfalls in this field and use the predictions only to help their general expert assessment of the results in front of them.
What has been surprising in the recent debate is the nature of the claims and counter-claims for “supremacy” when the absolute improvements in prediction “accuracy” are often less than the errors or normal variation expected between compounds measured on different instruments using different procedures, different solvents or concentrations.
As with most analytical techniques, the volume of reference data from which to draw hard and fast rules is negligible compared to the breadth of chemistry which a technique may have to span. Different strategies for 1H NMR spectra prediction have different strengths and weaknesses, but no particular approach dominates. The most common algorithms used for NMR prediction either draw on rules generated by studying the peer-reviewed literature values for chemical shifts or are reference database-based, where measured spectra and their assigned two-dimensional or three-dimensional chemical structures are the source knowledge base for training systems such as neural networks. I recently heard of an approach to 1H NMR prediction which seems to actually try for an obvious improvement.
Parameterisation approach
One approach which seems to deliver good results is parameterisation using functional group identification and information on the 3D structure of the compound to be predicted. The functional groups are identified and treated separately. In the work carried out by Professor Ray Abraham at Liverpool University, UK, a range of compounds with fixed geometry is selected and the 3D structures are retrieved using ab initio or molecular mechanics (MM) calculations. The associated 1H NMR data is then retrieved and assigned, with the parameters being varied to reproduce the experimental data most closely. This parameter set is then used as the prediction base for new chemical structures.
Currently, 20 different functional groups are used in this approach where good data is available. For some functional groups such as four-membered heterocyclic rings, azo-silicon and phosphorous compounds as well as charged compounds are not addressed and predictions for chemical structures containing these functional groups may be inaccurate. Professor Abraham has already contributed to this journal with an excellent overview of the CHARGE program and 1H NMR spectral prediction in general. Included in his article were some illuminating examples of where 1H NMR prediction had reached at that time.2
Additive increment rule-based approach
The long-standing rule-based approach of Professor Ernö Pretsch of the ETH Zurich, Switzerland, is widely used for 1H NMR and other spectra prediction software packages.3 This approach draws on years of analysing reference chemical shift values from the peer-reviewed literature, assigning a core value and functions by modifying a “base” proton chemical shift value for a substructure in a particular environment by various amounts depending on the location of neighbouring functional groups. The tables of chemical shift values and increments on which this approach is based was originally made available in book form, before being digitised and available for an extremely rapid computerised approach to 1H NMR spectra prediction.
Problems
In the Abraham article cited above, a clear description of the prediction problem is provided. A 1H NMR chemical shift value is not only dependent on the immediate connectivity, there is also a component which is due to longer range effects which are not capable of being modelled by 2D chemical structure drawings and require a 3D knowledge of the molecule in the matrix in which the spectrum is to be measured.2 The change from 2D to 3D molecular descriptors has reportedly already improved the prediction capabilities for the Pretsch approach on the Wiley 1H NMR reference database described below from around 0.30 ppm to 0.21 ppm.
It is worth remembering that you could have a superb quality reference database of 10,000 similar compounds of the class of molecule which you are trying to predict—and the best software in the world may deliver poor prediction accuracy due to a conformational difference only seen in your query molecule.
Interesting observation and a new strategy
Now what triggered the latest attempt at a step-change in prediction accuracy was the interesting observation that different prediction strategies actually go wrong (or worse) for different protons in the same molecule. Put more simply, it can often be the case that the approaches may all have an average error in their prediction approaching 0.3 ppm—somewhat worse than the 0.1–0.2 ppm requirement desired by scientists—but that different individual protons may be being predicted substantially better.
So why not try to identify the specific strengths and weaknesses of the different approaches and produce a combined prediction where the algorithm selected for a specific proton would depend on the experience gained on where each system was strongest.
Now, taking a 90,000 reference 1H NMR spectra available from Wiley yields around 1.1 million chemical shift values of non-exchangeable protons (not effected by concentration, solvent etc.), almost all measured in DMSO. The average prediction quality for the different approaches as well as 2D and 3D values for the Pretsch approach is given in Table 1.
Table 1. Prediction error for ~1.1 million 1H NMR chemical shift values predicted against the 90,000 Wiley 1H NMR reference database.
1H NMR prediction approach | Average prediction error |
Abraham (inherently 3D) | 0.28 ppm |
Pretsch 2D | 0.30 ppm |
Pretsch 3D | 0.21 ppm |
Professor Wolfgang Robien at the University of Vienna, Austria, has significant experience in looking for the “best” approach for 13C NMR prediction and his tools were able to work on the 1H NMR problem. Now if we were prescient and able to pick the best prediction approach by hand then the average prediction error dropped to an astonishing 0.14 ppm! Unfortunately, hindsight is a wonderful thing but we are required to draw up rules which select the “best” approach for compounds with no reference data where we do not have the luxury of comparing the predicted values with a reference quality measurement.
Initial automation of the selection of the most appropriate approach was carried out by Mike Wainright at Modgraph Consultants using 31 different chemical environments and the 2D Pretsch data set to classify the protons to be predicted. This yielded an improved average prediction error of 0.21 ppm from the 0.30 and 0.28 of the individual approaches. Almost at the target of 0.2 ppm.
More recently, two substantial improvements have been tested. Not only is the 3D Pretsch data set now available with the substantially improved prediction capability, but Wolfgang Robien has used 6300 one-bond HOSE codes as the selection criteria rather than the original 31 chemical environments. This has allowed the 0.2 ppm barrier to be breached for the first time for a dataset of such size and chemical diversity, yielding an average prediction error of 0.18 ppm (see Figure 1).
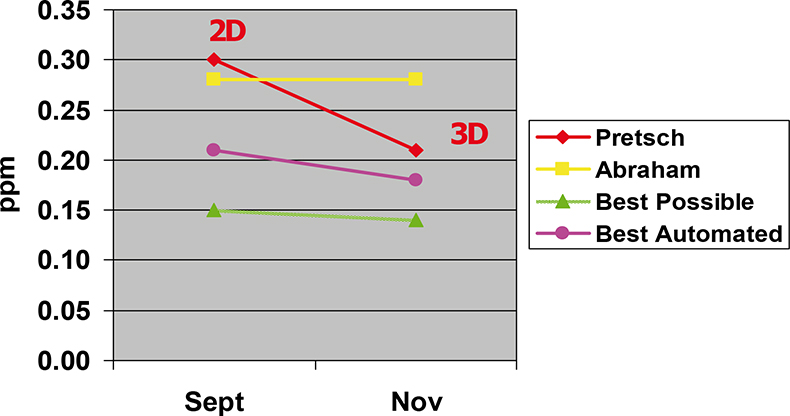
Figure 1. Diagram showing the improvements in average 1H NMR prediction error achievable by automatically combining the best prediction approach.
Conclusion
Clearly the combined approach is capable of producing significantly improved proton NMR predictions for the data set upon which it has been tested down to below the 0.2 ppm target error. As this data set is quite broad in its coverage of chemistry it would be interesting to try the same approach on a large data set covering a specific related class of chemical compounds such as produced by a single pharmaceutical or chemical company. Health Warning: This study has been carried out using data from compounds dissolved in DMSO—to apply the predictions to help interpret spectra measured in CDCl3 it would, of course, be useful to have a substantial reference data set also measured in CDCl3!
An additional benefit which was observed was a substantial reduction in the number of “outliers” where the predicted spectra are exceptionally poor.
The statistical analyses have also been fed back to the two groups and will hopefully provide a useful input for indentifying weaknesses and suggesting possible improvements.
This work has currently found its way into two commercial products, NMRPredict from Modgraph Consultants (see http://www.modgraph.co.uk/product_nmr.htm) from version 3.10.3 as well as integrated into the Mestrelab Research SL MestreNova product (see http://www.mestrec.com).
References
- Originally available in book form in several languages. Latest English edition E. Pretsch, P. Bühlmann, Ch. Affolter, Structure Determination of Organic Compounds: Tables of Spectral Data, 3rd completely revised and enlarged English Edn. Springer-Verlag, Berlin (2000). See also E. Pretsch, G. Tóth, M.E. Munk and M. Badertscher, Computer-Aided Structure Elucidation: Spectra Interpretation and Structure Generation. Wiley-VCH, Weinheim (2002). https://doi.org/10.1007/978-3-662-04201-4
- Raymond J. Abraham and Mehdi Mobli, “The prediction of 1H NMR chemical shifts in organic compounds”, Spectrosc. Europe 16(4), 16–22 (2004). https://www.spectroscopyeurope.com/article/prediction-1h-nmr-chemical-sh...
- R. Bürgin Schaller and E. Pretsch, “A computer program for the automatic estimation of 1H-NMR chemical shifts”, Anal. Chim. Acta 290, 295–302 (1994). https://doi.org/10.1016/0003-2670(94)80116-9